The BioBB REST API¶
The BioBB REST API allows the execution of the BioExcel Building Blocks in a remote server.
Documentation¶
For an extense documentation section, please go to the BioBB REST API website help.
Settings¶
Auxiliar libraries used¶
- requests: Requests allows you to send organic, grass-fed HTTP/1.1 requests, without the need for manual labor.
- nb_conda_kernels: Enables a Jupyter Notebook or JupyterLab application in one conda environment to access kernels for Python, R, and other languages found in other environments.
- nglview: Jupyter/IPython widget to interactively view molecular structures and trajectories in notebooks.
- ipywidgets: Interactive HTML widgets for Jupyter notebooks and the IPython kernel.
- plotly: Python interactive graphing library integrated in Jupyter notebooks.
Conda Installation and Launch¶
git clone https://github.com/bioexcel/biobb_REST_API_documentation.git
cd biobb_REST_API_documentation
conda env create -f conda_env/environment.yml
conda activate biobb_REST_API_documentation
jupyter-nbextension enable --py --user widgetsnbextension
jupyter-nbextension enable --py --user nglview
jupyter-notebook biobb_REST_API_documentation/notebooks/biobb_REST_API_documentation.ipynb
Behaviour¶
The BioBB REST API works as an asynchronous launcher of jobs, as these jobs can last from a few seconds to several minutes, there are some steps that must be performed for having the complete results of every tool.
BioExcel Building Blocks are structured in packages and tools. Every call to the BioBB REST API executes one single tool and returns the output file(s) related to this specific tool.
Tools information¶
List of packages¶
In order to get a complete list of available packages, we must do a GET request to the following endpoint:
https://mmb.irbbarcelona.org/biobb-api/rest/v1/launch
This endpoint returns a JSON HTTP response with status 200. More information in the BioBB REST API Documentation section.
List of tools¶
If there is need for a list of tools for a single package, we must do a GET request to the following endpoint:
https://mmb.irbbarcelona.org/biobb-api/rest/v1/launch/{package}
This endpoint returns a JSON HTTP response with status 200 or a 404 status if the package id is incorrect. More information in the BioBB REST API Documentation section.
Tool’s properties¶
If there is only need for the information of a single tool, we must do a GET request to the following endpoint:
https://mmb.irbbarcelona.org/biobb-api/rest/v1/launch/{package}/{tool}
This endpoint returns a JSON HTTP response with status 200 or a 404 status if the package id and / or the tool id are incorrect. The reason for failure should be detailed in the JSON response. More information in the BioBB REST API Documentation section.
Launch tool¶
For launching a tool, we must do a POST request to the following endpoint:
https://mmb.irbbarcelona.org/biobb-api/rest/v1/launch/{package}/{tool}
In the body of this POST request, we must add the file(s) needed as input (included the properties config file in JSON or YAML format) and the name for the output(s). The detailed list of inputs and outputs with its respectives properties can be found in the GET request of this same endpoint.
This endpoint returns a JSON HTTP response with the following possible status:
303: The job has been successfully launched and the user must save the token provided and follow to the next endpoint (defined in the same JSON response)404: There was some error launching the tool. The reason for failure should be detailed in the JSON response.500: The job has been launched, but some internal server error has occurred during the execution.
More information for a generic call in the BioBB REST API Documentation section. The documentation for all the tools is available in the BioBB REST API Tools Documentation section. Interactive examples for all the tools are available in the BioBB REST API Tools Execution section.
Retrieve status¶
If the previous endpoint returned a 303 status, we must do a GET request to the following endpoint providing the given token in the path:
https://mmb.irbbarcelona.org/biobb-api/rest/v1/retrieve/status/{token}
This endpoint checks the state of the job and returns a JSON HTTP response with the following possible status:
200: The job has finished successfully and in the JSON response we can found a list of output files generated by the job with its correspondent id for retrieving them on the next endpoint (defined in the same JSON message).202: The job is still running.404: Token incorrect, job unexisting or expired.500: Some internal server error has occurred during the execution.
More information in the BioBB REST API Documentation section.
Retrieve data¶
Once the previous endpoint returns a 200 status, the output file(s) are ready for its retrieval, so we must do a GET request to the following endpoint providing the given file id in the path:
https://mmb.irbbarcelona.org/biobb-api/rest/v1/retrieve/data/{id}
This endpoint returns the requested file with a 200 status or a 404 status if the provided id is incorrect, the file doesn’t exist or it has expired. More information in the BioBB REST API Documentation section.
Note that if we have executed a job that returns multiple output files, a call to this endpoint must be done for each of the output files generated by the job.
Sample files¶
The BioBB REST API provides sample files for most of the inputs and outputs of each tool. Files can be accessed thought the whole BioBB REST API hierarchical range.
All sample files¶
In order to download all the sample files, we must do a GET request to the following endpoint:
https://mmb.irbbarcelona.org/biobb-api/rest/v1/sample
This endpoint returns the requested file with a 200 status. More information in the BioBB REST API Documentation section.
Package sample files¶
In order to download all the sample files of a package, we must do a GET request to the following endpoint:
https://mmb.irbbarcelona.org/biobb-api/rest/v1/sample/{package}
This endpoint returns the requested file with a 200 status or a 404 status if the package id is incorrect. More information in the BioBB REST API Documentation section.
Tool sample files¶
In order to download all the sample files of a tool, we must do a GET request to the following endpoint:
https://mmb.irbbarcelona.org/biobb-api/rest/v1/sample/{package}/{tool}
This endpoint returns the requested file with a 200 status or a 404 status if the package id and / or the tool id are incorrect. The reason for failure should be detailed in the JSON response. More information in the BioBB REST API Documentation section.
Single sample file¶
In order to download a single sample file, we must do a GET request to the following endpoint:
https://mmb.irbbarcelona.org/biobb-api/rest/v1/sample/{package}/{tool}/{id}
This endpoint returns the requested file with a 200 status or a 404 status if the package id and / or the tool id and / or the file id are incorrect. The reason for failure should be detailed in the JSON response. More information in the BioBB REST API Documentation section.
Examples¶
Below we will do calls to all the previously defined endpoints and define some functions for make easier the connection to the BioBB REST API through Jupyter Notebook.
First off, we will import the Python requests and json library and set the root URI for the BioBB REST API.
import requests
import json
apiURL = "https://mmb.irbbarcelona.org/biobb-api/rest/v1/"
Tools information¶
Definition of simple GET / POST request functions and a class Response:
# Class for returning response status and json content of a requested URL
class Response:
def __init__(self, status, json):
self.status = status
self.json = json
# Perform GET request
def get_data(url):
r = requests.get(url)
return Response(r.status_code, json.loads(r.text))
# Perform POST request
def post_data(url, d, f):
r = requests.post(url, data = d, files = f)
return Response(r.status_code, json.loads(r.text))
List of packages¶
For more information about this endpoint, please visit the BioBB REST API Documentation section.
Endpoint¶
GET https://mmb.irbbarcelona.org/biobb-api/rest/v1/launch
Code¶
url = apiURL + 'launch'
response = get_data(url)
print(json.dumps(response.json, indent=2))
{
"packages": [ ... ]
}
List of tools from a specific package¶
For more information about this endpoint, please visit the BioBB REST API Documentation section.
Endpoint¶
GET https://mmb.irbbarcelona.org/biobb-api/rest/v1/launch/{package}
Code¶
package = 'biobb_analysis'
url = apiURL + 'launch/' + package
response = get_data(url)
print(json.dumps(response.json, indent=2))
{
"id": "biobb_analysis",
"tools": [ ... ]
}
Tool’s properties¶
For more information about this endpoint, please visit the BioBB REST API Documentation section.
Endpoint¶
GET https://mmb.irbbarcelona.org/biobb-api/rest/v1/launch/{package}/{tool}
Code¶
package = 'biobb_analysis'
tool = 'cpptraj_average'
url = apiURL + 'launch/' + package + '/' + tool
response = get_data(url)
print(json.dumps(response.json, indent=2))
{
"id": "cpptraj_average",
"description": "Calculates a structure average of a given cpptraj compatible trajectory.",
"arguments": [
{
"id": "config",
"required": false,
"description": "Configuration file for the cpptraj_average tool",
"filetype": "input",
"sample": "https://raw.githubusercontent.com/bioexcel/biobb_analysis/master/biobb_analysis/test/data/config/config_cpptraj_average.json",
"formats": [
".*\\.json$",
".*\\.yml$"
]
},
{
"id": "input_top_path",
"required": true,
"description": "Path to the input structure or topology file",
"filetype": "input",
"sample": "https://github.com/bioexcel/biobb_analysis/raw/master/biobb_analysis/test/data/ambertools/cpptraj.parm.top",
"formats": [
".*\\.top$",
".*\\.pdb$",
".*\\.prmtop$",
".*\\.parmtop$",
".*\\.zip$"
]
},
{
"id": "input_traj_path",
"required": true,
"description": "Path to the input trajectory to be processed",
"filetype": "input",
"sample": "https://github.com/bioexcel/biobb_analysis/raw/master/biobb_analysis/test/data/ambertools/cpptraj.traj.dcd",
"formats": [
".*\\.crd$",
".*\\.cdf$",
".*\\.netcdf$",
".*\\.restart$",
".*\\.ncrestart$",
".*\\.restartnc$",
".*\\.dcd$",
".*\\.charmm$",
".*\\.cor$",
".*\\.pdb$",
".*\\.mol2$",
".*\\.trr$",
".*\\.gro$",
".*\\.binpos$",
".*\\.xtc$",
".*\\.cif$",
".*\\.arc$",
".*\\.sqm$",
".*\\.sdf$",
".*\\.conflib$"
]
},
{
"id": "output_cpptraj_path",
"required": true,
"description": "Path to the output processed structure",
"filetype": "output",
"sample": "https://github.com/bioexcel/biobb_analysis/raw/master/biobb_analysis/test/reference/ambertools/ref_cpptraj.average.pdb",
"formats": [
".*\\.crd$",
".*\\.netcdf$",
".*\\.rst7$",
".*\\.ncrst$",
".*\\.dcd$",
".*\\.pdb$",
".*\\.mol2$",
".*\\.binpos$",
".*\\.trr$",
".*\\.xtc$",
".*\\.sqm$"
]
}
]
}
Launch tool¶
For more information about this endpoint, please visit the BioBB REST API Documentation section. The documentation for all the tools is available in the BioBB REST API Tools Documentation section. Interactive examples for all the tools are available in the BioBB REST API Tools Execution section.
Definition of functions needed for launch a job:
from io import BytesIO
from pathlib import Path
# Function used for encode python dictionary to JSON file
def encode_config(data):
jsonData = json.dumps(data)
binaryData = jsonData.encode()
return BytesIO(binaryData)
# Launch job
def launch_job(url, **kwargs):
data = {}
files = {}
# Fill data (output paths) and files (input files) objects
for key, value in kwargs.items():
# Inputs / Outputs
if type(value) is str:
if key.startswith('input'):
files[key] = (value, open(value, 'rb'))
elif key.startswith('output'):
data[key] = value
elif Path(value).is_file():
files[key] = (value, open(value, 'rb'))
# Properties (in case properties are provided as a dictionary instead of a file)
if type(value) is dict:
files['config'] = ('prop.json', encode_config(value))
# Request URL with data and files
response = post_data(url, data, files)
# Print REST API response
print(json.dumps(response.json, indent=2))
# Save token if status == 303
if response.status == 303:
token = response.json['token']
return token
Hereafter we will launch a job on biobb_analysis.cpptraj_average tool with the provided files/ in the files folder of this same repository. The response is a JSON with the status code, the state of the job, a message and a token for checking the job status.
Launch job with a YAML file config¶
File config¶
properties:
in_parameters:
start: 1
end: -1
step: 1
mask: c-alpha
out_parameters:
format: pdb
Endpoint¶
POST https://mmb.irbbarcelona.org/biobb-api/rest/v1/launch/{package}/{tool}
Code¶
The function below sends POST data and files to the {package}/{tool} endpoint. The config properties are sent as a YAML file.
The response is a JSON with the status code, the state of the job, a message and a token that will be used for checking the job status in the next step.
# Launch BioBB on REST API with YAML config file
token = launch_job(url = apiURL + 'launch/biobb_analysis/cpptraj_average',
config = 'files/config.yml',
input_top_path = 'files/cpptraj.parm.top',
input_traj_path = 'files/cpptraj.traj.dcd',
output_cpptraj_path = 'output.cpptraj.average.pdb')
{
"code": 303,
"state": "RUNNING",
"message": "The requested job has has been successfully launched, please go to /retrieve/status/{token} for checking job status.",
"token": "fe2805760eeeec0d5b8a34fbc40aa6c2a2d68c7ba1663cccb88659b1e149c898a414bbc04e37bb73efc725b7a29de2a93ffb55e6ef85cd6467f3d62a06ea5bfa"
}
Launch job with a JSON file config¶
File config:
{
"in_parameters": {
"start": 1,
"end": -1,
"step": 1,
"mask": "c-alpha"
},
"out_parameters": {
"format": "pdb"
}
}
Endpoint¶
POST https://mmb.irbbarcelona.org/biobb-api/rest/v1/launch/{package}/{tool}
Code¶
The function below sends POST data and files to the {package}/{tool} endpoint. The config properties are sent as a JSON file.
The response is a JSON with the status code, the state of the job, a message and a token that will be used for checking the job status in the next step.
# Launch BioBB on REST API with JSON config file
token = launch_job(url = apiURL + 'launch/biobb_analysis/cpptraj_average',
config = 'files/config.json',
input_top_path = 'files/cpptraj.parm.top',
input_traj_path = 'files/cpptraj.traj.dcd',
output_cpptraj_path = 'output.cpptraj.average.pdb')
{
"code": 303,
"state": "RUNNING",
"message": "The requested job has has been successfully launched, please go to /retrieve/status/{token} for checking job status.",
"token": "84ab5ef63d82ab3fa4f120532949905d83f6aff65f101cb1ed5fdd5f05acb00421ddc4560098f877f26a96972a8ea8521ab222a0bb78a5ffa9d213c0ab2618c9"
}
Launch job with a python dictionary config¶
Endpoint¶
POST https://mmb.irbbarcelona.org/biobb-api/rest/v1/launch/{package}/{tool}
Code¶
The function below sends POST data and files to the {package}/{tool} endpoint. The config properties are sent as a python dictionary embedded in the code.
The response is a JSON with the status code, the state of the job, a message and a token that will be used for checking the job status in the next step.
# Launch BioBB on REST API with JSON config file
prop = {
"in_parameters" : {
"start": 1,
"end": -1,
"step": 1,
"mask": "c-alpha"
},
"out_parameters" : {
"format": "pdb"
}
}
token = launch_job(url = apiURL + 'launch/biobb_analysis/cpptraj_average',
config = prop,
input_top_path = 'files/cpptraj.parm.top',
input_traj_path = 'files/cpptraj.traj.dcd',
output_cpptraj_path = 'output.cpptraj.average.pdb')
{
"code": 303,
"state": "RUNNING",
"message": "The requested job has has been successfully launched, please go to /retrieve/status/{token} for checking job status.",
"token": "98013d74bef397d5498db3eb1008e5e136702d63903b6ea0cb5a2db44c4a4e0adbcd1ce9999915acd90c444f8749880c052185bbbfc747c1ebc7d67d6d2c84c8"
}
Retrieve status¶
For more information about this endpoint, please visit the BioBB REST API Documentation section.
Definition of functions needed for retrieve the status of a job:
import datetime
from time import sleep
# Checks status until a provided "ok" status is returned by the response
def check_status(url, ok, error):
counter = 0
while True:
if counter < 10: slp = 1
if counter >= 10 and counter < 60: slp = 10
if counter >= 60: slp = 60
counter = counter + slp
sleep(slp)
r = requests.get(url)
if r.status_code == ok or r.status_code == error:
return counter
break
# Function that checks the status and parses the reponse JSON for saving the output files in a list
def check_job(token, apiURL):
# define retrieve status URL
url = apiURL + 'retrieve/status/' + token
# check status until job has finished
counter = check_status(url, 200, 500)
# Get content when status = 200
response = get_data(url)
# Save id for the generated output_files
if response.status == 200:
out_files = []
for outf in response.json['output_files']:
item = { 'id': outf['id'], 'name': outf['name'] }
out_files.append(item)
# Print REST API response
print("Total elapsed time: %s" % str(datetime.timedelta(seconds=counter)))
print("REST API JSON response:")
print(json.dumps(response.json, indent=4))
if response.status == 200:
return out_files
else: return None
Endpoint¶
GET https://mmb.irbbarcelona.org/biobb-api/rest/v1/retrieve/status/{token}
Code¶
The function below checks the status of a job and awaits until the response status is 200. The response is a JSON with the status code, the state of the job, a message, a list with all the generated output files and the date of the expiration of these files. Additionally, the function also provides the elapsed time since the job has been launched until it has finished.
# Check job status
out_files = check_job(token, apiURL)
Total elapsed time: 0:00:20
REST API JSON response:
{
"code": 200,
"state": "FINISHED",
"message": "The requested job has finished successfully, please go to /retrieve/data/{id} for each output_files.",
"output_files": [
{
"id": "5e42837a40fe75.05757111",
"name": "output.cpptraj.average.pdb",
"size": 77397,
"mimetype": "text/plain"
}
],
"expiration": "February 13, 2020 00:00 GMT+0000"
}
Retrieve data¶
For more information about this endpoint, please visit the BioBB REST API Documentation section.
Definition of functions needed for retrieve the output file(s) generated by a job:
# Downloads to disk a file from a given URL
def get_file(url, filename):
r = requests.get(url, allow_redirects=True)
file = open(filename,'wb')
file.write(r.content)
file.close()
# Retrieves all the files provided in the out_files list
def retrieve_data(out_files, apiURL):
if not out_files:
return "No files provided"
for outf in out_files:
get_file(apiURL + 'retrieve/data/' + outf['id'], outf['name'])
Endpoint¶
GET https://mmb.irbbarcelona.org/biobb-api/rest/v1/retrieve/data/{id}
Code¶
The function below makes a single call to the retrieve/data endpoint for each output file got in the retrieve/status endpoint and save the generated file(s) to disk.
# Save generated file(s) to disk
retrieve_data(out_files, apiURL)
Practical cases¶
Now we will execute some Bioexcel Building Blocks through the BioBB REST API and with the results we will do some interactions with other python libraries such as plotly or nglview.
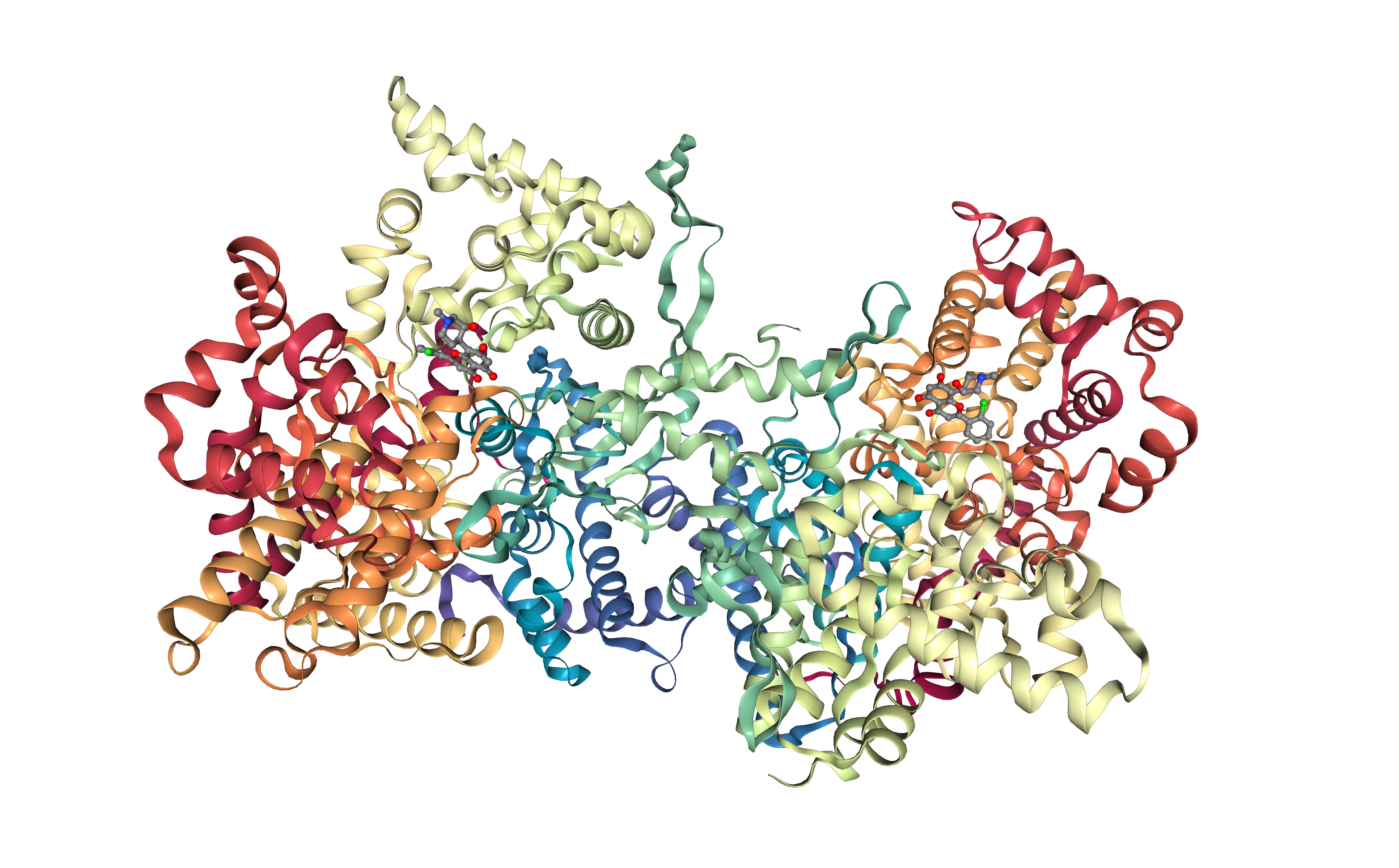
Example 1: download PDB file from RSCB database¶
Launch the biobb_io.pdb job that downloads a PDB file from the RSCB database:
# Downloading desired PDB file
# Create properties dict and inputs/outputs
downloaded_pdb = '3EBP.pdb'
prop = {
'pdb_code': '3EBP',
'filter': False
}
# Launch bb on REST API
token = launch_job(url = apiURL + 'launch/biobb_io/pdb',
config = prop,
output_pdb_path = downloaded_pdb)
{
"code": 303,
"state": "RUNNING",
"message": "The requested job has has been successfully launched, please go to /retrieve/status/{token} for checking job status.",
"token": "af60d733db949a71167f3aa6a7a793fc520b5a4176b57a770bed4798654a79be2a47b81e6a77de4eb285de84f9b768b119004f0bfbbe4be9e5ff1ffe31b81fd9"
}
# Check job status
out_files = check_job(token, apiURL)
Total elapsed time: 0:00:06
REST API JSON response:
{
"code": 200,
"state": "FINISHED",
"message": "The requested job has finished successfully, please go to /retrieve/data/{id} for each output_files.",
"output_files": [
{
"id": "5e428389eeafa3.49051362",
"name": "3EBP.pdb",
"size": 609120,
"mimetype": "text/plain"
}
],
"expiration": "February 13, 2020 00:00 GMT+0000"
}
# Save generated file to disk
retrieve_data(out_files, apiURL)
Visualize downloaded PDB in NGLView:
import nglview
# Show protein
view = nglview.show_structure_file(downloaded_pdb)
view.add_representation(repr_type='ball+stick', selection='het')
view._remote_call('setSize', target='Widget', args=['','600px'])
view
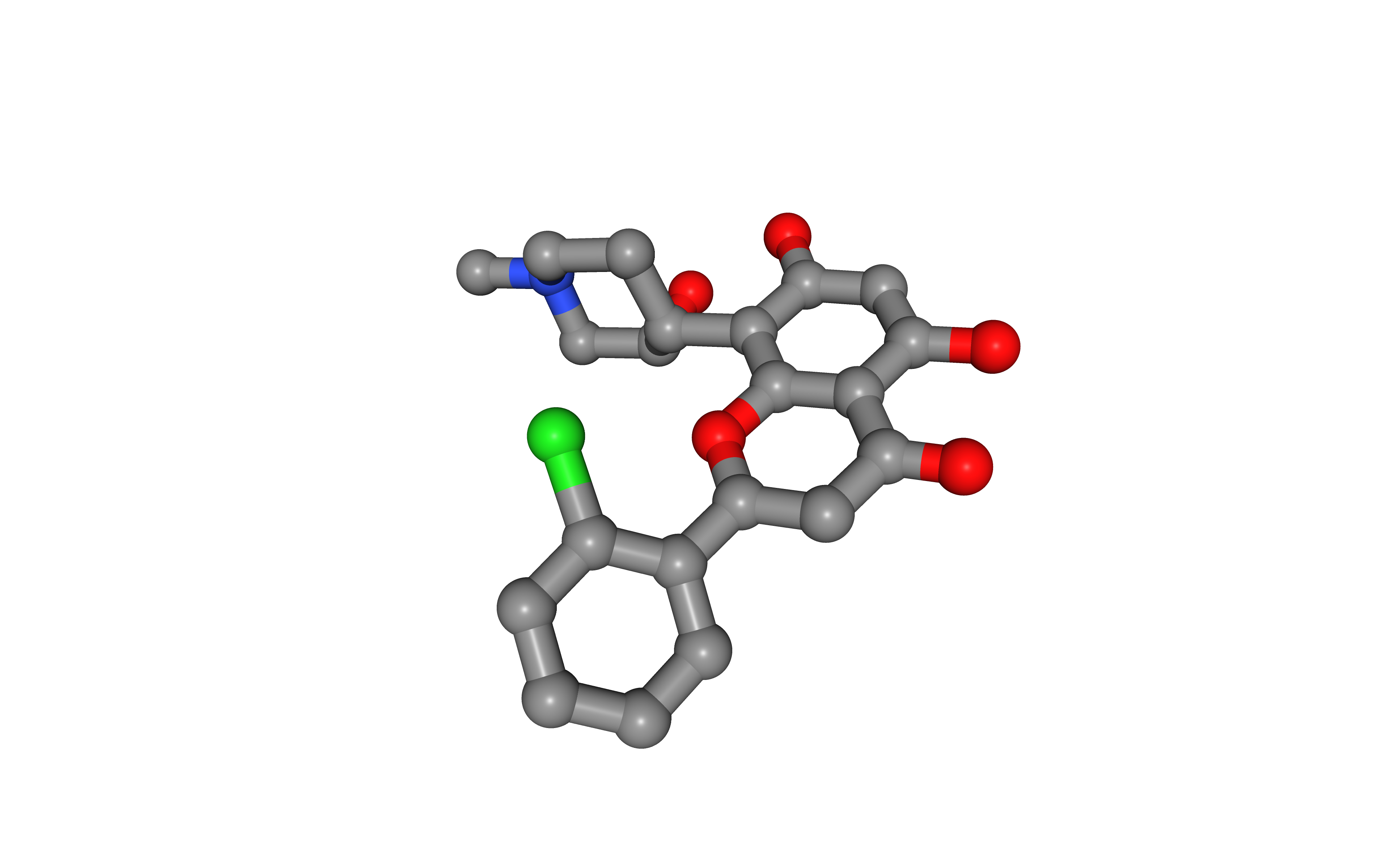
Example 2: extract heteroatom from a given structure¶
Launch the biobb_structure_utils.extract_heteroatoms job that extracts a heteroatom from a PDB file.
# Extracting heteroatom from a given structure
# Create properties dict and inputs/outputs
heteroatom = 'CPB.pdb'
prop = {
'heteroatoms': [{
'name': 'CPB'
}]
}
# Launch bb on REST API
token = launch_job(url = apiURL + 'launch/biobb_structure_utils/extract_heteroatoms',
config = prop,
input_structure_path = downloaded_pdb,
output_heteroatom_path = heteroatom)
{
"code": 303,
"state": "RUNNING",
"message": "The requested job has has been successfully launched, please go to /retrieve/status/{token} for checking job status.",
"token": "740c9ac30767ba996e445e4ed05c151ee903fed2234c0cc7ace6ec3ba4e1fa8bdcd5a3c6835c7f1038530eb81c4cc319674f235cf55b38863903181dce09a8d1"
}
# Check job status
out_files = check_job(token, apiURL)
Total elapsed time: 0:00:20
REST API JSON response:
{
"code": 200,
"state": "FINISHED",
"message": "The requested job has finished successfully, please go to /retrieve/data/{id} for each output_files.",
"output_files": [
{
"id": "5e4283986555a0.86371712",
"name": "CPB.pdb",
"size": 2268,
"mimetype": "text/plain"
}
],
"expiration": "February 13, 2020 00:00 GMT+0000"
}
# Save generated file to disk
retrieve_data(out_files, apiURL)
Visualize generated extracted heteroatom in NGLView:
# Show protein
view = nglview.show_structure_file(heteroatom)
view.add_representation(repr_type='ball+stick', selection='het')
view._remote_call('setSize', target='Widget', args=['','600px'])
view
Example 3: extract energy components from a given GROMACS energy file¶
# GMXEnergy: Getting system energy by time
# Create prop dict and inputs/outputs
output_min_ene_xvg ='file_min_ene.xvg'
output_min_edr = 'files/1AKI_min.edr'
prop = {
'terms': ["Potential"]
}
# Launch bb on REST API
token = launch_job(url = apiURL + 'launch/biobb_analysis/gmx_energy',
config = prop,
input_energy_path = output_min_edr,
output_xvg_path = output_min_ene_xvg)
{
"code": 303,
"state": "RUNNING",
"message": "The requested job has has been successfully launched, please go to /retrieve/status/{token} for checking job status.",
"token": "170e8e2645d179eaa40e2de652f3e6dec909ef1df4642526ba789ed21806bab917bb4ce7f9fb730dfe635155f52ac9f3869c4429afcc767bd190f01337a8a718"
}
# Check job status
out_files = check_job(token, apiURL)
Total elapsed time: 0:00:08
REST API JSON response:
{
"code": 200,
"state": "FINISHED",
"message": "The requested job has finished successfully, please go to /retrieve/data/{id} for each output_files.",
"output_files": [
{
"id": "5e4283a6c70143.38956052",
"name": "file_min_ene.xvg",
"size": 54143,
"mimetype": "text/plain"
}
],
"expiration": "February 13, 2020 00:00 GMT+0000"
}
# Save generated file to disk
retrieve_data(out_files, apiURL)
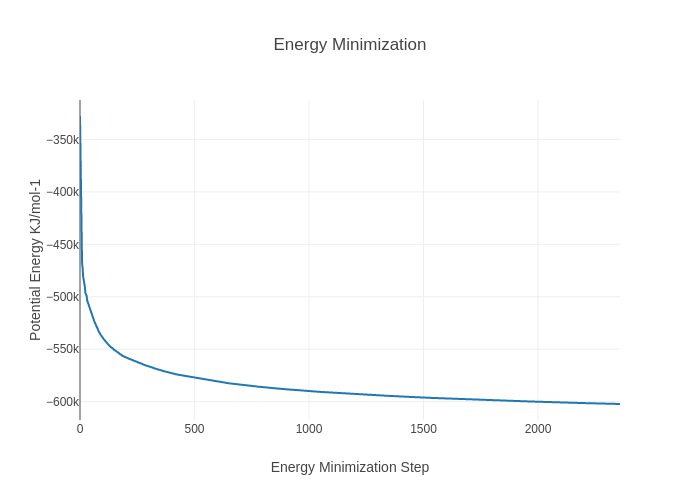
Visualize generated energy file in plotly:
import plotly
import plotly.graph_objs as go
#Read data from file and filter energy values higher than 1000 Kj/mol^-1
with open(output_min_ene_xvg,'r') as energy_file:
x,y = map(
list,
zip(*[
(float(line.split()[0]),float(line.split()[1]))
for line in energy_file
if not line.startswith(("#","@"))
if float(line.split()[1]) < 1000
])
)
plotly.offline.init_notebook_mode(connected=True)
fig = {
"data": [go.Scatter(x=x, y=y)],
"layout": go.Layout(title="Energy Minimization",
xaxis=dict(title = "Energy Minimization Step"),
yaxis=dict(title = "Potential Energy KJ/mol-1")
)
}
plotly.offline.iplot(fig)